محتويات المقال
مرض الثلاسيميا
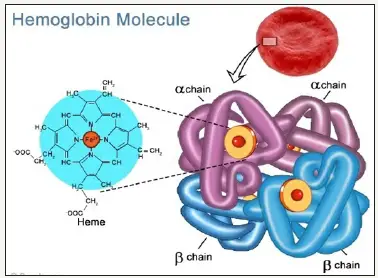
الثلاسيميا (Thalassemia) أو أنيميا البحر المتوسط هو مرض وراثي في الدم أي ينتقل عبر الأجيال، يتميز بعدم قدرة الجسم على إنتاج ما يكفي من الهيموجلوبين، وهو مكون رئيسي لخلايا الدم الحمراء.
عندما لا تحتوي خلايا الدم الحمراء في الجسم على ما يكفي من الهيموجلوبين، فإنها لا تعمل بشكل فعال وتعيش لفترات زمنية أقصر، مما يؤدي إلى انخفاض عدد خلايا الدم الحمراء السليمة التي تتدفق عبر مجرى الدم.
يمكن أن يكون فقر الدم طفيفًا أو شديدًا عند الأشخاص المصابين بالثلاسيميا، ويمكن أن يتسبب فقر الدم الحاد في تلف الأعضاء وحتى الموت.
اسباب مرض الثلاسيميا
الثلاسيميا هي حالة وراثية، مما يعني أن أحد الوالدين على الأقل يجب أن يكون حاملاً للمرض، أو حدوث طفرة وراثية أو فقدان شرائح جينية حرجة معينة.
الثلاسيميا هي مجموعة متنوعة من الأمراض الوراثية الناتجة عن انخفاض إنتاج الهيموغلوبين ألفا أو سلسلة بيتا. الهيموجلوبين هو المكون الحامل للأكسجين في خلايا الدم الحمراء، ويتكون من بروتينين أحدهما ألفا والآخر بيتا.
عندما لا ينتج الجسم ما يكفي من أحد هذين البروتينين، لا تتطور خلايا الدم الحمراء بشكل صحيح ولا يمكنها حمل ما يكفي من الأكسجين، مما يؤدي إلى فقر الدم الذي يمكن أن يبدأ في الطفولة ويستمر مدى الحياة.
انواع مرض الثلاسيميا
ثلاسيميا ألفا وبيتا ثلاسيميا هما النوعان الأساسيان من الثلاسيميا، يؤثر كل منهما على جزء مختلف من الهيموجلوبين (البروتين الموجود في خلايا الدم الحمراء الذي يحمل الأكسجين).
إن بيتا غلوبين وألفا غلوبين مكونان متميزان (وحدات فرعية) من الهيموغلوبين، يقوم جين HBB بتوجيه إنتاج بيتا غلوبين، بينما تقوم جينات HBA1 و HBA2 بتوجيه إنتاج alpha globin. يتم تناقل كل من هذه الجينات في نسختين، واحدة من الأم والأخرى من الأب.
ينتج ثلاسيميا بيتا عن تغيرات (طفرات) في جين HBB، مما يؤدي إلى انخفاض مستويات بيتا جلوبين. وينتج نقص غلوبين ألفا من فقدان (حذف) بعض أو كل جينات HBA1 أو HBA2، مما يؤدي إلى الإصابة بالثلاسيميا ألفا.
حامل مرض الثلاسيميا
يتأثر إنتاج الهيموغلوبين بأربعة جينات وتسبب مشاكل هذه الجينات مرض الثلاسيميا، ولكن إذا كان هناك خلل واحد فقط من الجينات الأربعة، يُقال إن الشخص مصاب بالثلاسيميا وليس لديه أعراض.
الثلاسيميا مرض وراثي، وتنتقل الجينات الشاذة من الآباء إلى أطفالهم. يمكن لأي شخص أن يكون حامل لمرض الثلاسيميا عندما يتلقى أحد الجينات من أحد الوالدين.
لا يعاني الأشخاص الحاملين لمرض الثلاسيميا أي من الأعراض المرتبطة بالثلاسيميا، مثل فقر الدم.
مرض الثلاسيميا والزواج
يتم توريث الثلاسيميا في نمط صبغي جسدي متنحي في معظم الحالات. ومع ذلك، يمكن أن تكون الوراثة معقدة لأن العديد من الجينات يمكن أن تؤثر على إنتاج الهيموجلوبين.
في معظم الأشخاص المصابين بالثلاسيميا بيتا، توجد طفرات في نسختين من جين HBB في كل خلية. يُعرف والدان الشخص المصاب بالناقلين لأن لكل منهما نسخة واحدة متحولة من الجين.
على الرغم من أن بعض حاملي الثلاسيميا بيتا يصابون بفقر دم معتدل، إلا أن معظم حاملي الثلاسيميا لا تظهر عليهم أي علامات أو أعراض للمرض.
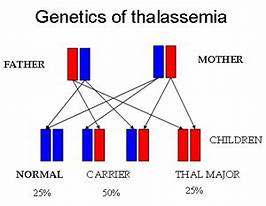
عندما يكون اثنان من الآباء حاملي الاضطرابات الصبغية المتنحية، فإن كل طفل لديه فرصة بنسبة 25٪ (1 من 4) للإصابة بهذا المرض، وفرصة بنسبة 50٪ (1 من كل 2) أن يكون حاملاً للمرض مثل كلا الوالدين، وفرصة 25٪ في عدم الإصابة بهذا المرض. وجود الشرط وليس كونه ناقل.
ترتبط وراثة ألفا ثلاسيميا بحقيقة أن الحالة مرتبطة بطفرات في جينين منفصلين هما (HBA1 و HBA2)، تحتوي كل خلية على نسختين من جين HBA1 ونسختين من جين HBA2، نسخة واحدة من كل جين موروثة من الأم والأخرى من الأب.
إن الآباء الذين فقدوا نسخة جينية واحدة على الأقل أطفالهم معرضون لخطر الإصابة بالثلاسيميا ألفا. ومع ذلك، فإن عدد نسخ الجينات المفقودة (المحذوفة) من الجينات HBA1 و HBA2 هي التي تحدد المخاطر وشدة المرض لكل طفل.
اعراض مرض الثلاسيميا عند الكبار
نظرًا لأن عند الإصابة بالثلاسيميا جسمك يحتوي على عدد أقل من خلايا الدم الحمراء، فقد تظهر عليك أعراض فقر الدم وقد تواجه أيضًا:
- الدوخة.
- ضيق التنفس.
- تسارع ضربات القلب.
- الصداع.
- تشنجات في الساق.
- صعوبة في التركيز.
- شحوبة البشرة.
يُعَدْ نخاع العظم ذلك المكون الإسفنجي الداكن في وسط العظام، هو الموقع الأساسي لإنتاج خلايا الدم. نظرًا لأن نخاع العظم يعمل بجهد أكبر من المعتاد؛ لإنتاج خلايا دم حمراء إضافية فقد يتمدد.
يؤدي هذا إلى تمدد عظامك مما قد يؤدي إلى شدها وجعلها أرق وأكثر عرضة للكسر.
الطحال هو عضو آخر في الجسم ينتج الدم، إنه تحت ضلوعك السفلية على الجانب الأيسر من بطنك. يؤدي الطحال مجموعة متنوعة من الوظائف من أهمها تصفية الدم ومراقبة الدم بحثًا عن عدوى معينة.
عندما تكون مصابًا بالثلاسيميا، ينمو الطحال في الحجم حيث يحاول إنتاج خلايا الدم، لكن لا يمكنها تصفية الدم أو التحقق من العدوى ومكافحتها.
يعاني الأشخاص المصابون بالثلاسيميا من “نقص المناعة”، مما يشير إلى أن عناصر دفاعات الجسم المضادة للعدوى لا تعمل.
عندما يكون جهازك المناعي ضعيفًا، يكون من السهل العدوى بالأمراض، وقد تحتاج إلى حماية إضافية، مثل لقاحات الإنفلونزا والتحصينات الأخرى.
اقرأ أيضْا الامراض المناعية.
اعراض مرض الثلاسيميا عند الأطفال
قد لا تظهر أي أعراض على الأطفال الذين يعانون من أنواع أقل حدة من الاضطراب، بينما يمكن أن يعاني الأطفال المصابون بالأنواع الأكثر شدة من مرض الثلاسيميا من:
- شحوب الجلد بشكل ملحوظ.
- اصفرار الجلد والعينين (اليرقان).
- انتفاخ البطن بسبب تضخم الطحال أو الكبد.
- عظام الوجه بارزة جدا.
- توقف النمو.
- ممارسة التعصب.
- النفخة القلبية (أصوات غير طبيعية في القلب بسبب فقر الدم).
تشخيص مرض الثلاسيميا
يتوفر الاختبار الجيني للجينات HBB و HBA1 و HBA2، المعروف أنها تسبب مرض الثلاسيميا، إذا كانت الطفرات المسببة للمرض في الأسرة معروفة.
إن اختبار الناقل للأقارب المعرضين للخطر واختبار ما قبل الولادة من الخيارات أيضًا.
سجل الاختبارات الجينية (GTR) هو قاعدة بيانات موحدة على الإنترنت للمعلومات الجينية، ويتضمن أيضًا معلومات عن ثلاسيميا ألفا والاختبار الجيني للثلاسيميا بيتا.
يجب على المرضى والمستهلكين الذين لديهم أسئلة معينة بخصوص الاختبار الجيني التحدث مع طبيب أو خبير في علم الوراثة.
عادةً ما يتم تشخيص مرض الثلاسيميا عن طريق فحص حديثي الولادة يتلقاها كل طفل. يمكن لهذا الفحص الكشف عن أكثر أشكال الثلاسيميا شيوعًا وشدة.
إذا لم يتم اكتشافه خلال فحص حديثي الولادة، فيمكن اكتشافه أثناء الفحص الروتيني لفقر الدم من قبل مقدم الرعاية الأولية الخاص بك عندما يبلغ طفلك من عام إلى عامين.
الوقاية من مرض الثلاسيميا
من الصعب للغاية تجنب مرض الثلاسيميا لأنه ينتقل من الآباء إلى الأبناء. إذا كان لديك أنت أو شريكك فرد من أفراد الأسرة مصاب بالثلاسيميا.
أو إذا كان لكلاكما أقارب من البلدان التي ينتشر فيها مرض الثلاسيميا، يمكنك استشارة مستشار وراثي؛ لتحديد مخاطر انتقال مرض الثلاسيميا إلى أطفالك.
علاج مرض الثلاسيميا
تختلف خيارات العلاج لمرض الثلاسيميا حسب نوع الحالة وشدتها، فلا تظهر أعراض على الأشخاص الحاملين لمرض ثلاسيميا ألفا أو بيتا، ربما لن يحتاجوا سوى القليل من العلاج.
بالنسبة للحالات الخفيفة والشديدة من مرض الثلاسيميا، يستخدم الأطباء ثلاثة علاجات شائعة، من بين العلاجات المتاحة:
- عمليات نقل الدم
قد تحتاج إلى عمليات نقل دم إذا كنت تعاني من بيتا ثلاسيميا وسيطة، وإذا كنت مصابًا بالثلاسيميا بيتا الكبرى (Cooley’s anemia).
فستحتاج على الأرجح إلى عمليات نقل الدم بشكل متكرر (غالبًا كل أسبوعين إلى أربعة أسابيع). ستحافظ عمليات نقل الدم هذه على مستويات الهيموجلوبين وخلايا الدم الحمراء لديك تحت السيطرة.
- علاج استخلاب الحديد (ke-LAY-shun)
الهيموغلوبين هو بروتين غني بالحديد يوجد في خلايا الدم الحمراء. نتيجة لذلك، قد يؤدي نقل الدم المنتظم إلى تراكم الحديد في الدم.
يشار إلى هذا بالحمل الزائد للحديد. يضر الكبد والقلب وأعضاء الجسم الأخرى، يستخدم الأطباء العلاج باستخلاب الحديد للتخلص من الحديد الزائد من الجسم لمنع هذا الضرر.
- مكملات حمض الفوليك
حمض الفوليك هو فيتامين ب الذي يساعد في بناء خلايا الدم الحمراء الصحية. قد يوصي طبيبك بمكملات حمض الفوليك بالإضافة إلى العلاجات السابقة.
تم اكتشاف علاجات أخرى أو ما زالت في طور الاختبار، على الرغم من استخدامها بشكل أقل تكرارًا مثل: زرع الخلايا الجذعية في الدم والنخاع العظمي.
اقرأ أيضْا التبرع بالدم ..حقائق وإرشادات.
References
- https://www.cdc.gov/ncbddd/thalassemia/facts.html
- https://rarediseases.info.nih.gov/diseases/7756/thalassemia
- https://www.nhlbi.nih.gov/health-topics/thalassemias
- https://www.nicklauschildrens.org/conditions/thalassemia-trait
- https://www.cdc.gov/ncbddd/thalassemia/treatment.html
- https://www.childrens.com/specialties-services/conditions/thalassemia

